
Pharmaceutical translation services deliver regulatory-compliant translation of drug labels, patient information leaflets (PILs), clinical study reports, CTDs, and submission documents for EMA, FDA, and global health authorities. Accurate pharmaceutical translation requires subject-matter-qualified translators, validated termbases, back-translation for specific instruments, and a documented quality workflow aligned with ICH, GMP, and ISO 17100 standards.
Pharmaceutical translation operates at the intersection of language accuracy, regulatory compliance, and patient safety. A mistranslated dosage instruction, contraindication, or adverse event description can delay market authorisation, trigger regulatory queries, or create real clinical risk.
Life sciences buyers therefore prioritise translator qualifications, terminology control, and audit-ready workflows over cost. Circle Translations delivers native pharmaceutical translators with scientific degrees, two-stage review, validated termbases, multilingual DTP for labels and PILs, and full NDA-backed confidentiality for all regulatory and clinical documentation.
Pharmaceutical Translation Document Types: From Clinical Development to Post-Market Surveillance

Pharmaceutical translation spans the full product lifecycle, from clinical research and regulatory submission to commercial launch and post-market pharmacovigilance, each stage requiring precise, standardised translation aligned with health authority expectations.
| Document Type | Regulatory Context | Translation Direction | Key Accuracy Requirements | Health Authority Reference |
| Drug label and labelling artwork | EU Annex II / FDA 21 CFR Part 201 | EN → EU languages; EN for FDA | Dosage, contraindications, storage — zero ambiguity | EMA; FDA |
| Patient Information Leaflet (PIL) | EU Directive 2001/83/EC Art. 63 | EN → EU languages; EN/ES US | Plain-language readability; patient comprehension | EMA QRD; FDA |
| SmPC | EU Directive 2001/83/EC Annex I | EN → EU languages | Clinical accuracy; QRD alignment | EMA |
| CTD | ICH M4 | Mixed (Module-dependent) | Section accuracy; MedDRA consistency | ICH; EMA; FDA |
| CSR | ICH E3 | EN + summaries localised | Clinical/statistical precision | ICH |
| IB | ICH E6 (GCP) | EN + local | Safety + preclinical accuracy | ICH |
| ICF | ICH E6; 21 CFR Part 50 | Local languages | Patient clarity; ethics approval accuracy | FDA; IRB |
| PSUR / PBRER | ICH E2C(R2) | EN + local summaries | Pharmacovigilance terminology | EMA PRAC |
| REMS | FDAAA 2007 | EN + ES | Safety communication completeness | FDA |
| PV / ADR reports | EU 536/2014 | Local | MedDRA coding accuracy | EMA; FDA |
| IFU | ISO 15223 | All markets | Safety symbols; warnings | EU MDR; FDA |
| COAs / PROs | FDA / EMA guidance | EN → local | Linguistic validation required | FDA; EMA |
Drug Label and PIL Translation: EMA QRD Template Compliance and EU Readability Requirements
Drug labels and PILs require strict EMA QRD template compliance and validated patient readability across all target languages.
QRD templates define mandatory section headings and approved phrasing in 24 EU languages, eliminating free translation. INNs remain unchanged, while dosage values and decimal conventions must match local standards (e.g., 2.5 mg → 2,5 mg).
Article 59 requires readability testing per language, meaning translations must be adapted for patient comprehension, not literal accuracy. Multilingual DTP ensures layout integrity, managing 20–35% text expansion and preserving diacritics in the final artwork.
CTD Translation for EMA and FDA Submissions: Module Structure, ICH Alignment, and MedDRA Terminology
CTD translation requires module-specific handling, strict ICH alignment, and exact MedDRA terminology usage across all clinical data. Module 1 is localised per authority, while Modules 2–5 remain largely in English unless required by markets like Japan or China.
Every section must follow ICH M4, E3, and related guidelines without paraphrasing. MedDRA terms must match approved target-language versions exactly, any deviation creates regulatory inconsistencies and potential query letters.
Cross-module consistency is mandatory, as mismatched terminology signals data integrity risk during review.
Linguistic Validation for COAs and PROs: Back-Translation, Cognitive Debriefing, and FDA/EMA Requirements
COA and PRO translation requires full linguistic validation, not standard translation, to prove conceptual equivalence across languages.
The process includes 7 stages: dual forward translation, reconciliation, dual back-translation, review, harmonisation, cognitive debriefing with 5–10 native participants, and final documentation.
Regulators require evidence that translated instruments measure the same clinical constructs as the original.
Without this documented validation workflow, PRO data risks rejection in regulatory submissions, particularly under FDA guidance.
Pharmacovigilance Translation: PSUR, Adverse Event Reports, and MedDRA-Aligned Safety Narratives
Pharmacovigilance translation demands exact MedDRA alignment, precise causality language, and zero ambiguity in safety reporting.
PSUR/PBRER documents must accurately reflect benefit–risk evaluations under ICH E2C(R2), while ICSRs require correct MedDRA LLT/PT coding to support signal detection.
Standard causality categories (e.g., probable, possible) must be used exactly as defined. Any deviation in adverse event terminology or SAE definitions can distort safety signals and trigger regulatory scrutiny, making precision non-negotiable.
Pharmaceutical Translation Quality Framework: ISO 17100, GMP Alignment, and Terminology Control

Pharmaceutical translation quality is a documented, audit-ready system combining ISO 17100 processes, GMP documentation control, and strict terminology governance to meet regulatory inspection standards.
| Quality Standard | What Does It Require in Pharmaceutical Translation | How It Is Implemented |
| ISO 17100:2015 | Qualified translator + independent reviser; documented workflow; terminology control | Subject-matter translators; mandatory second review; full audit trail |
| GMP (EU Annex 11 / 21 CFR Part 211) | Version control; traceability, and documented changes | Version-controlled files; full change history; named contributors |
| ICH Q10 | Lifecycle quality management; CAPA; risk-based control | Translation QA reviews; corrective actions; risk scoring by document |
| EMA QRD compliance | Approved terminology; fixed structure | QRD templates embedded in termbases; automated checks |
| Back-translation protocol | Independent reverse translation for validation | Independent back-translators; discrepancy reports |
| Linguistic validation | 7-stage validated workflow (FDA/EMA) | Full validation process with cognitive debriefing |
| Audit trail | Inspection-ready documentation | Complete records; translator qualifications; QC logs |
Pharmaceutical Termbase Management: INN Consistency, MedDRA Alignment, and Controlled Vocabulary
Pharmaceutical termbase management enforces absolute consistency across INNs, MedDRA terminology, and regulatory vocabulary to prevent classification errors and regulatory queries. Pharmaceutical terminology does not allow synonym variation. Each term carries a fixed regulatory meaning.
INNs and ATC codes remain untranslated, while MedDRA Preferred Terms (PTs) and Lowest Level Terms (LLTs) must match approved target-language versions exactly.
A structured termbase includes active substances, QRD phrases, dosage language, safety terminology, and company-specific naming conventions, all enforced in CAT tools with version control and deviation tracking.
Back-Translation in Pharmaceutical Contexts: When It’s Required, How It Works, and What the Output Must Contain
Back-translation validates conceptual accuracy by independently translating the target text back into the source language and comparing discrepancies against the original. It is mandatory for COA/PRO linguistic validation and is often applied to high-risk documents like informed consent forms or safety-critical labelling.
The process requires an independent back-translator with no access to the source, followed by a structured comparison report and discrepancy resolution log.
Regulatory validity depends on independence—non-independent back-translation invalidates the entire quality assurance process.
Pharmaceutical Translation Quality Audit Trail: What Inspection-Ready Documentation Looks Like
An inspection-ready pharmaceutical translation audit trail documents every decision, contributor, and quality check from project brief to final delivery.
Required records include translator and reviser qualifications, termbase version history, QA reports, and tracked changes showing all revisions. GMP-aligned systems ensure full traceability, enabling rapid retrieval during EMA or FDA inspections.
Missing audit documentation signals non-compliance, even if the translation itself is accurate, making documentation as critical as linguistic quality.
Regulatory Translation Requirements by Market: EMA, FDA, ICH, and National Health Authorities
Pharmaceutical translation requirements vary by market, with each authority defining specific language obligations, document sets, and validation standards that directly affect approval timelines and compliance risk.
| Market / Authority | Language Requirements | Key Translation Documents | Back-Translation Required | Certification / Legalisation | Reference Standard |
| EU — EMA | Module 1 + product info in all EU languages | SmPC, PIL, labels | COA/PRO only | No | Directive 2001/83/EC; QRD |
| EU — National | National language per country | SmPC, PIL, labels | COA/PRO | Varies | National agencies |
| US — FDA | English and Spanish in practice for patients | PI, MedGuide, REMS | COA/PRO | No (standard) | 21 CFR Part 201 |
| Japan — PMDA | Japanese mandatory | Full CTD; PI | Sometimes | Yes | PMDA guidelines |
| China — NMPA | Simplified Chinese | Full CTD; labels | COA/PRO | Sometimes notarised | NMPA CTD |
| Brazil — ANVISA | Brazilian Portuguese | Dossier; bula | Not required | Sometimes notarised | ANVISA RDC |
| Australia — TGA | English | PI; CMI | COA/PRO | No | TGA guidance |
| WHO PQ | English + local languages | PIL; dossier | COA | No | WHO PQ |
EMA Translation Requirements: QRD Templates, 24-Language Label Obligations, and Article 59 Readability
EMA requires translation into up to 24 EU languages, strict QRD template compliance, and validated patient readability under Article 59. Product information (SmPC, PIL, labels) must use QRD-approved wording—no free translation allowed.
Each language version must follow the latest QRD template and pass readability testing with ~10 target users. This ensures patient comprehension, not just linguistic accuracy.
The shift to electronic product information (ePI) adds structured XML/FHIR translation requirements, increasing technical complexity.
FDA Pharmaceutical Translation: Package Insert Requirements, REMS, and the Spanish Labelling Landscape
FDA requires English labelling but increasingly expects Spanish patient-facing materials for real-world compliance and REMS programmes.
The Package Insert (PI) follows strict PLR formatting under 21 CFR 201, while Medication Guides and REMS materials often require Spanish versions for patient accessibility.
Clinical trial materials (e.g., informed consent forms) must be translated for non-English study sites. Unlike EMA, translation is not centralised but driven by patient access and safety communication needs.
Pharmaceutical Translation for Emerging Markets: ANVISA, NMPA, PMDA, and WHO Prequalification Requirements
Emerging markets require full local-language submissions with stricter translation scope and higher compliance risk due to limited regulatory tolerance for errors. Brazil mandates Brazilian Portuguese dossiers and bula translations, often with notarisation.
China requires full Simplified Chinese CTD submissions with consistent pharmacopoeia terminology. Japan demands complete Japanese CTD translation and adherence to J-MedDRA.
WHO Prequalification adds multilingual PIL requirements across diverse markets, increasing linguistic and operational complexity.
Commission Pharmaceutical Translation With Audit-Ready Quality

Pharmaceutical translation is a regulatory risk—Circle Translations delivers audit-ready output aligned with EMA, FDA, and ICH standards.
✓ Pharmaceutical-specialist translators with verified credentials
✓ ISO 17100 two-stage review on every project
✓ EMA QRD + MedDRA + INN terminology control
✓ GMP-compliant audit trail (fully documented, inspection-ready)
✓ Back-translation and linguistic validation available
✓ NDA-secured, confidential handling
Send your documents and get a qualified proposal within 1 business hour →
Pharmaceutical Translation Services — Frequently Asked Questions
What qualifications should a pharmaceutical translator hold?
A pharmaceutical translator must have a life sciences degree (e.g., pharmacy, biochemistry), professional translation training, and proven experience with EMA, FDA, and ICH frameworks. They must also use MedDRA terminology correctly and work within CAT tools and termbases. For regulatory documents, always request translator CVs and confirm subject-matter expertise in the specific therapeutic area.
How much do pharmaceutical translation services cost?
Pharmaceutical translation typically costs $0.18–$0.35 per word, depending on document type and regulatory complexity. CTD modules, SmPCs, and PILs sit at the higher end, while linguistic validation ranges from $3,000–$15,000+ per language. DTP for labelling adds $500–$2,500+, and rush delivery increases cost by 25–50%.
What is the difference between pharmaceutical translation and medical translation?
Pharmaceutical translation focuses on drug development and regulatory documents, while medical translation covers broader healthcare content. Pharmaceutical work requires strict EMA/FDA/ICH compliance, MedDRA alignment, and documented QA processes, making it more regulated and technically demanding than general medical translation.
Is machine translation acceptable for pharmaceutical regulatory documents?
Machine translation is not acceptable for regulatory or patient-facing pharmaceutical documents. It lacks terminology precision, audit trails, and compliance with regulatory standards. MTPE is only suitable for low-risk internal content and should never be used for CTDs, labels, PILs, or clinical data.
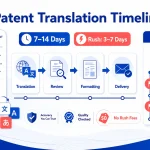
How long does it take to translate a drug label or PIL into multiple EU languages?
A full EU translation (up to 24 languages) typically takes 10–20 business days, depending on scope and DTP complexity. Single-language projects take 2–5 days, while accelerated timelines (5–7 days) require parallel workflows and pre-planned project management.
What is ICH E6 and why does it matter for pharmaceutical translation?
ICH E6 (GCP) governs clinical trial conduct and requires accurate translation of participant-facing documents. It mandates native-language informed consent forms and full documentation of translation processes within the Trial Master File, making compliance essential for trial approval.
Can Circle Translations manage multi-language pharmaceutical projects?
Yes—multi-language pharmaceutical translation programmes are managed with centralised workflows and shared termbases across all languages. Parallel translation teams ensure consistency for large EMA submissions, while a dedicated project manager maintains alignment across all language versions.
What is the difference between a certified and a notarised pharmaceutical translation?
Certified translation confirms accuracy via a signed statement, while notarised translation verifies the translator’s identity through a notary. Certification is common for regulatory submissions, while notarisation is required in specific markets like Brazil or certain national authorities.
How is confidentiality handled in pharmaceutical translation?
Pharmaceutical translation requires strict NDA-based confidentiality with controlled access to sensitive data. Files are handled on a need-to-know basis, excluded from MT training, and supported by data processing agreements for GDPR compliance, ensuring full protection of proprietary and clinical data.